{kind=link}
Tokyo Medical and Dental University (TMDU) researchers have discovered that patients with 18q deletion syndrome can experience both cellular and humoral immunodeficiency

Credit: Department of Child Health and Development, TMDU
Tokyo Medical and Dental University (TMDU) researchers have discovered that patients with 18q deletion syndrome can experience both cellular and humoral immunodeficiency
Tokyo, Japan – Chromosome 18q deletion (18q del) syndromeis a rare genetic condition disorder, affecting approximately 1 in 40,000 to 55,000 individuals, caused by the deletion of genetic material on the long arm of chromosome 18. This genetic anomaly disrupts normal growth and development, and critically, can impair the immune system’s functionality. Patients with 18q del syndrome often exhibit humoral immunodeficiency or a common variable immunodeficiency (CVID)-like phenotype, characterized by low levels of immunoglobulins (antibodies) in the blood, compromising the body’s ability to effectively combat infections.
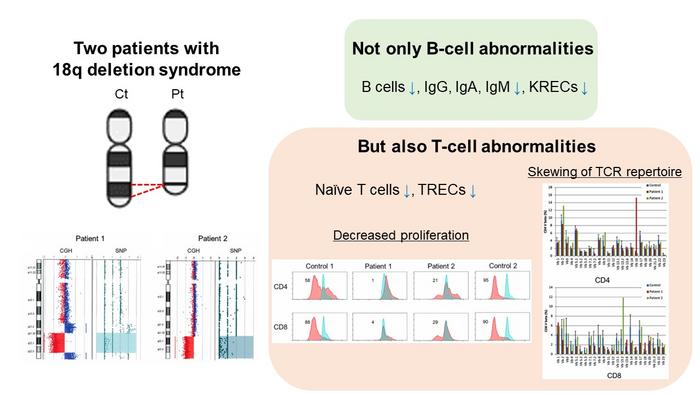
Now, however, in a study published recently in the Journal of Clinical Immunology, researchers from Tokyo Medical and Dental University (TMDU) and Kagoshima University have identified a previously undocumented manifestation among patients with 18q del syndrome: late-onset combined immunodeficiency (LOCID), affecting both B and T cells. This novel finding underscores the critical importance of routinely assessing the functionality of both B and T cells in individuals with 18q del syndrome.
Elaborating further on this novel finding, Professor Kanegane says, “In this study, we came across two patients with chromosome 18q del syndrome presenting with LOCID, which, to the best of our knowledge, has not yet been reported in patients with the syndrome.”
Patient 1 was a 29-year-old man diagnosed with 18q del syndrome. Despite initially having few infections, he developed Pneumocystis pneumonia (PCP). Array-based comparative genomic hybridization (CGH) analysis showed a deletion in the 18q21.32-q22.3 chromosome region.
Patient 2 was a 48-year-old woman who had not been previously diagnosed with 18q del syndrome. However, she was diagnosed with granulomatous lymphadenitis, and a biopsy of her lymph nodes revealed a loss of 18q21.33-qter.
Both patients exhibited hypogammaglobulinemia, characterized by abnormally low levels of immunoglobulins (IgG, IgA, IgM, and IgE). Patient 1’s serum immunoglobulin levels were significantly below normal ranges. He reported IgG of 188 mg/dL (normal: 870–1,700 mg/dL), IgA of 105 mg/dL (normal: 110–410 mg/dL), IgM of 26 mg/dL (normal: 33–190 mg/dL), and IgE of <5 IU/mL (normal: 232 IU/mL). His CD4+ T cells had a decreased percentage of naïve T cells, accounting for only 3.58% of the total CD3+CD4+ cell population. Moreover, his T-cell receptor excision circles (TREC) levels and Ig κ-deleting recombination excision circle (KREC) levels were extremely low at 25.27 copies/105 cells (normal: > 565 copies/105 cells) and 93.36 copies/105 cells (normal: ≥ 456 copies/105 cells) respectively, indicating poor T-cell production.
Similar conditions were noted for patient 2, who reported IgG of 8 mg/dL, IgA of 9 mg/dL, IgM of 131 mg/dL, and IgE of 0.3 IU/mL. Her CD4+ T cells and naïve CD4+ T cells were depleted, with naïve T cells accounting for only 6% of the CD3+CD4+ cell population. Her TREC levels were 0 copies/105 cells, and her KREC levels were 11.4 copies/105 cells.
Importantly, CD4+ and CD8+ T cells failed to divide in response to phytohemagglutinin (PHA) stimulation, indicating severe functional impairment of T cells in both the patients.
Based on their immune profiles and clinical history, both of them were diagnosed with LOCID, a condition where both humoral (antibody-mediated) and cell-mediated immune responses were impaired, making them highly susceptible to infections.
This novel finding is significant, as Dr. Tomomasa, the co-authored of this study, states, “While cases involving deletion of the same region as those of the two patients presented in this study have been reported earlier, patients with 18q del syndrome developing LOCID have never been reported. We speculate that these patients simply have not yet developed LOCID or that they might not have been adequately assessed for it.”
On the basis of these results, the researchers recommend annual testing for both cellular and humoral immunity in patients with 18q del syndrome. This proactive approach can allow for the early detection of combined immune deficiencies, facilitating timely interventions and personalized treatment strategies. Ultimately, such regular monitoring can significantly improve clinical outcomes and enhance the quality of life for individuals diagnosed with 18q del syndrome.
###
The article, “18q Deletion Syndrome Presenting with Late-Onset Combined Immunodeficiency,” was published in the Journal of Clinical Immunology at DOI: 10.1007/s10875-024-01751-4
Journal
Journal of Clinical Immunology
Article Title
18q Deletion Syndrome Presenting with Late-Onset Combined Immunodeficiency