Amyotrophic lateral sclerosis (ALS) represents one of the most devastating neurodegenerative conditions, relentlessly eroding motor neuron function and leading to debilitating loss of muscle control. Typically, patients face respiratory failure within three to five years following diagnosis, underscoring the urgent need for innovative therapeutic interventions. In a groundbreaking study led by the Institute for Neurosciences (IN), a collaborative center of Miguel Hernández University (UMH) and the Spanish National Research Council (CSIC), researchers have unveiled a critical deficiency in chaperone-mediated autophagy (CMA) within motor neurons of ALS patients. This discovery opens promising avenues for targeting cellular protein clearance pathways as a strategy to halt or slow ALS progression.
The study, recently published in Acta Neuropathologica Communications, goes beyond previous understandings of autophagy in ALS by focusing specifically on CMA, a selective mechanism by which cells degrade aberrant or damaged proteins. Unlike macroautophagy, which broadly targets cellular debris, CMA identifies and degrades specific protein cargos, a function essential to maintaining neuronal homeostasis. In ALS, where toxic aggregates of the RNA-binding protein TDP-43 accumulate abnormally in motor neurons, the failure of such precision clearance mechanisms could be a driver of cellular degeneration.
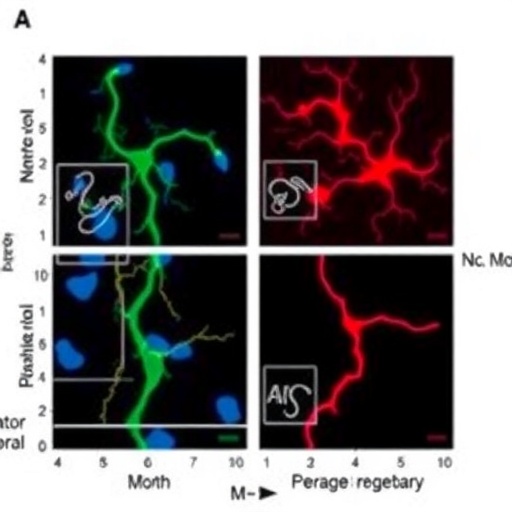
To delve into these molecular intricacies, researchers obtained post-mortem spinal cord tissues from ALS patients enrolled in clinical trials and from age-matched healthy donors. Employing advanced immunohistochemistry and immunofluorescence techniques, they quantified levels of LAMP2A, a lysosomal membrane receptor pivotal for substrate translocation in CMA. Strikingly, motor neurons from ALS patients exhibited a pronounced reduction in LAMP2A expression and activity compared to controls, pointing to a compromised CMA pathway as a consistent hallmark in the diseased state.
ALS predominantly affects motor neurons, specialized nerve cells responsible for initiating muscle contractions. In the majority of ALS cases, these neurons harbor cytoplasmic aggregates of TDP-43, a protein normally residing in the nucleus and involved in RNA metabolism. The aberrant cytoplasmic localization of TDP-43 is toxic, disrupting cellular function and viability. The researchers propose that a decline in CMA preferentially impairs the degradation of such proteins, facilitating their pathological accumulation. This observation challenges prior assumptions that generalized autophagy declines were solely responsible, highlighting CMA’s unique and indispensable role.
Professor Salvador Martínez, the laboratory director overseeing this research, emphasized that restoring CMA function may be pivotal for motor neuron survival. “Motor neurons require exceptionally high CMA activity to maintain proteostasis. When this system falters, as observed in ALS, these neurons become especially vulnerable, leading to the progressive neurodegeneration characteristic of the disease,” he explained. This insight underscores the therapeutic potential of pharmacologically or genetically modulating CMA to augment its protein-clearance capacity.
One of the most compelling aspects of this investigation is the direct demonstration of CMA dysfunction in human neuronal tissue, a feat rarely achievable in animal models. This human-centric approach affirms the biological relevance of CMA alterations and enhances translational prospects. The detailed cellular analyses reveal that the malfunction is not an incidental side effect but a targeted failure of protein clearance pathways specific to motor neuron populations impacted by ALS.
The scientific team’s multidisciplinary collaboration included experts from the UMH Sports Research Centre and the Pascual Parrilla Murcia Institute for Biosanitary Research, reflecting the integrated efforts required to tackle a disease of such complexity. They point out that this work was only feasible thanks to the invaluable donations of neural tissue by ALS patients and their families. Such altruistic contributions provide irreplaceable biological material essential for unraveling ALS mechanisms and testing novel hypotheses.
Mechanistically, chaperone-mediated autophagy involves the recognition of substrate proteins by cytosolic chaperones, which then transport these targets to lysosomal membranes bearing LAMP2A receptors. Following binding, substrates translocate into the lysosome for degradation, thus preventing toxic protein build-up. The discovery that LAMP2A expression is diminished in ALS motor neurons suggests that this critical gateway is impaired, obstructing the selective removal of pathological proteins like TDP-43 and potentially amplifying neurotoxicity.
This study’s findings pave the way for exploring CMA-enhancing therapies that could restore cellular balance in ALS-affected neurons. By developing small-molecule activators or gene therapies aimed at increasing LAMP2A levels or stabilizing chaperone function, researchers aim to reverse toxic protein accumulation. Although in the early stages, such strategies could transform the current landscape of ALS treatment, which remains largely supportive without disease-modifying options.
The researchers also underscore the necessity of further investigations to elucidate how CMA dysfunction interacts with other cellular degradation pathways and contributes to the complex ALS pathology. Synergistic therapeutic strategies combining CMA enhancement with modulation of other proteostatic mechanisms may hold the key to achieving meaningful clinical benefits. This integrative view deepens understanding of motor neuron biology under stress and neurodegenerative demands.
Funding for this pivotal work came from several prestigious sources, including the Spanish State Research Agency’s Severo Ochoa Excellence Programme, the Ministry of Science, Innovation and Universities, the Generalitat Valenciana’s Prometeo Programme, and the Instituto de Salud Carlos III’s Advanced Therapies Network (TERAV). Additionally, support from the Next Generation EU initiative and UMH’s Gregoria Ramos Gil Chair on ALS was instrumental in facilitating comprehensive research efforts.
In conclusion, this landmark study reveals that chaperone-mediated autophagy is critically impaired in spinal motor neurons affected by ALS, contributing to the accumulation of neurotoxic TDP-43 aggregates. This deficiency emerges as a compelling molecular target to develop therapeutic approaches capable of preserving motor neuron integrity and delaying disease progression. As scientists decode the complex interplay of cellular clearance systems, new hope arises for patients facing the relentless challenges of ALS.
Subject of Research: Human tissue samples
Article Title: Chaperone mediated autophagy is deficient in spinal motoneurons of ALS patients with TDP-43 proteinopathy
News Publication Date: 4-Feb-2026
Web References: http://dx.doi.org/10.1186/s40478-026-02238-6
Image Credits: Instituto de Neurociencias UMH CSIC
Keywords: Amyotrophic lateral sclerosis, ALS, motor neurons, TDP-43, chaperone-mediated autophagy, CMA, protein aggregation, neurodegeneration, LAMP2A, autophagy regulation, neurobiology

{kind=link}