In a groundbreaking study set to reshape our understanding of DYT1 dystonia, researchers have uncovered a novel pathological mechanism involving the dysregulation of nuclear Lamin B1. This discovery, published in Cell Death Discovery, offers unprecedented insights into how alterations within the nuclear lamina contribute to the onset and progression of this debilitating movement disorder. The findings not only illuminate the molecular underpinnings of DYT1 dystonia but also open new avenues for therapeutic intervention aimed at restoring nuclear integrity and protein homeostasis.
DYT1 dystonia, a hereditary neurological disorder characterized by involuntary muscle contractions and abnormal postures, has long puzzled scientists striving to decode its cellular origins. The current research by Duan, Sepehrimanesh, Hosain, and colleagues highlights Lamin B1, a crucial structural component of the nuclear envelope, as a central player whose dysregulation thickens the nuclear lamina, disrupting normal nuclear function. Lamin B1’s aberrant accumulation fortifies the lamina beyond its physiological parameters, precipitating a cascade of cellular dysfunctions that culminate in the dystonic phenotype.
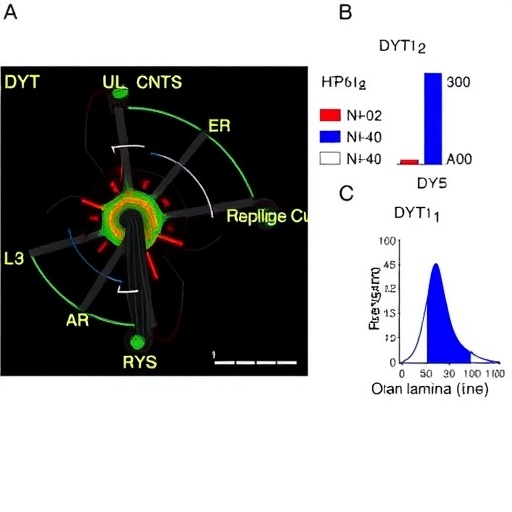
At the heart of the nuclear envelope lies the lamina, a meshwork of intermediate filament proteins providing mechanical support and regulating essential nuclear processes like DNA replication and transcription. Lamin B1, integral to forming this network, ensures nuclear stability and the faithful execution of genetic programs. However, in DYT1 dystonia, the precise balance of Lamin B1 is perturbed, as demonstrated by the significant lamina thickening reported in affected cells. This abnormal nuclear stiffening is hypothesized to impede normal nuclear-cytoplasmic communication and genomic regulation, thereby contributing to neuronal dysfunction.
The study delves further into the consequences of Lamin B1 dysregulation, revealing its impact on the 14-3-3 protein family, key regulators of intracellular signaling pathways. These adaptor proteins modulate a wide range of cellular processes, including apoptosis, cell cycle control, and signal transduction. The thickened nuclear lamina influences 14-3-3 protein distribution and function, disrupting their interactions and impairing their regulatory capacity. This disruption represents a critical nexus through which nuclear architecture aberrations translate into widespread cellular disturbances.
Employing advanced imaging techniques and biophysical analyses, the authors meticulously quantified nuclear morphology changes correlating with Lamin B1 accumulation. The thickened lamina was not merely a passive structural anomaly but a dynamic contributor to compromised nuclear mechanics. These alterations affect nuclear envelope plasticity and potentially hinder the trafficking of molecules essential for neuronal survival and adaptability, exacerbating disease etiology.
The researchers utilized patient-derived cellular models alongside animal systems genetically engineered to recapitulate DYT1 dystonia. These models confirmed that Lamin B1 dysregulation induced nuclear lamina thickening and 14-3-3 protein mislocalization, resulting in neuronal dysfunction consistent with dystonia pathophysiology. Crucially, interventions that modulated Lamin B1 expression or stabilized 14-3-3 protein interactions ameliorated some pathological phenotypes, underscoring the therapeutic potential of targeting this pathway.
Insightful biochemical analyses shed light on the molecular mechanisms linking Lamin B1 overload to 14-3-3 protein disruption. Altered post-translational modifications of 14-3-3 proteins, influenced by the structural changes of the nuclear lamina, were implicated in diminished protein binding affinity and altered signaling outcomes. These findings illustrate the intimate crosstalk between nuclear architecture and intracellular signaling networks, vital for maintaining neuronal health.
Furthermore, the study highlights the significance of nuclear elasticity in neuron function. Neurons, reliant on plastic nuclear properties to adapt to mechanical stresses and regulate gene expression, suffer profoundly when the nuclear lamina is pathologically stiffened. The elevated rigidity caused by Lamin B1 excess impairs nuclear mechanotransduction pathways, potentially triggering maladaptive cellular responses and contributing to dystonia’s progressive nature.
By dissecting the intracellular consequences of nuclear lamina abnormalities, the research underscores an emerging paradigm in neurodegenerative and movement disorders: structural nuclear components are not mere scaffolds but dynamic regulators whose perturbations have far-reaching effects. This paradigm shift promises to invigorate future studies exploring the nuclear lamina as a therapeutic target, inviting innovative strategies to restore both nuclear integrity and protein signaling homeostasis.
An exciting implication of this research lies in its potential to generalize across other laminopathies and neurological conditions featuring nuclear envelope defects. The mechanistic insights gained from DYT1 dystonia could catalyze cross-disciplinary investigations into how nuclear lamina alterations may drive pathogenesis in a spectrum of disorders, potentially revolutionizing approaches to diseases currently lacking effective treatments.
The study also raises intriguing questions about the regulation of nuclear lamin homeostasis under normal and pathological conditions. Understanding the molecular cues controlling Lamin B1 synthesis, degradation, and turnover will be essential for designing interventions that delicately recalibrate nuclear lamina composition without disrupting physiological functions. Strategies harnessing targeted proteostasis pathways may emerge as promising therapeutic modalities.
Moreover, the impact on 14-3-3 proteins emphasizes the interplay between nuclear structure and cytoplasmic signaling networks. Future research may explore whether similar regulatory disruptions occur in other adaptor protein families, broadening our comprehension of how nuclear envelope integrity governs cellular homeostasis. Such exploration holds promise for unraveling complex intracellular communication pathways perturbed in dystonia.
The clinical repercussions of these findings are profound. By identifying Lamin B1 as a modulatable factor in dystonia pathogenesis, the study furnishes a tangible target for drug development. Pharmacological agents or gene therapies aimed at reducing Lamin B1 levels or restoring 14-3-3 functionality could transform therapeutic landscapes, offering hope to patients enduring the relentless progression of DYT1 dystonia.
In conclusion, this pioneering study brings to light an intricate molecular ballet whereby dysregulated nuclear Lamin B1 thickens the nuclear lamina and disrupts critical 14-3-3 proteins, unraveling the cellular fabric underlying DYT1 dystonia. The amalgamation of structural cell biology and protein signaling revelations heralds a new era in understanding and ultimately countering this enigmatic disorder. As the scientific community embraces these insights, novel therapeutic horizons beckon, promising to redefine patient outcomes.
Subject of Research: Nuclear Lamin B1 dysregulation and its role in the pathogenesis of DYT1 dystonia.
Article Title: Dysregulated nuclear Lamin B1 in DYT1 dystonia thickens nuclear lamina and disrupts 14-3-3 proteins.
Article References:
Duan, Y., Sepehrimanesh, M., Hosain, M.A. et al. Dysregulated nuclear Lamin B1 in DYT1 dystonia thickens nuclear lamina and disrupts 14-3-3 proteins. Cell Death Discov. (2026). https://doi.org/10.1038/s41420-026-03090-2
Image Credits: AI Generated

{kind=link}