{kind=link}
Prostate cancer is the second most common cancer among men worldwide, following lung cancer. In the United States alone, nearly 300,000 new cases are diagnosed annually. While reducing testosterone and other male hormones can be an effective treatment for prostate cancer, this approach becomes ineffective once the disease progresses to metastatic castration-resistant prostate cancer (mCRPC). At this stage, the cancer advances quickly and becomes resistant to conventional hormonal therapies and chemotherapy.

Credit: Tomoya Uehara from Chiba University
Prostate cancer is the second most common cancer among men worldwide, following lung cancer. In the United States alone, nearly 300,000 new cases are diagnosed annually. While reducing testosterone and other male hormones can be an effective treatment for prostate cancer, this approach becomes ineffective once the disease progresses to metastatic castration-resistant prostate cancer (mCRPC). At this stage, the cancer advances quickly and becomes resistant to conventional hormonal therapies and chemotherapy.
A clever strategy for fighting mCRPC is to exploit the fact that these tumor cells usually overexpress a membrane protein called prostate-specific membrane antigen (PSMA). In particular, targeted alpha therapy involves attaching a radioactive atom like actinium-225 (225Ac) to a compound that binds strongly to PSMA. As the radioactive atom decays, it emits alpha particles that are harmful to nearby cells—in this case, tumor cells. However, given that the production of 225Ac is very low, scientists are looking for more viable alternatives.
In a recent study, a research team led by Professor Tomoya Uehara from Chiba University, Japan, developed a novel compound for the targeted alpha therapy of prostate cancer using a different alpha particle emitting radionuclide: astatine-211 (211At). Other members of the team included Hiroyuki Suzuki and Kento Kannaka from Chiba University, as well as Kazuhiro Takahashi from Fukushima Medical University. Their findings, which were published in Volume 9 of EJNMMI Radiopharmacy and Chemistry on June 17, 2024, address one of the main issues plaguing 211At-based compounds for targeted alpha therapy: deastatination.
Simply put, deastatination refers to the natural process by which enzymes in the body cleave the 211At atom from the whole compound, effectively splitting the therapeutic part from the PSMA-targeting part. This not only renders the drug unable to address the cancer itself, but also releases a radioactive payload to other tissues in the body, which can damage the liver, stomach, and kidneys.
To avoid this problem, the researchers turned to a chemical structure they had previously studied. “Recently, we developed a neopentyl derivative with two hydroxy groups, which we referred to as an ‘NpG structure,’ as a 211At-labeling moiety that could stably retain 211At in vivo,” explains Uehara, “Based on these past results, we hypothesized that the NpG structure could be used to design a variety of 211At-labeled PSMA-targeting derivatives.”
The team put their theory to the test by designing and synthesizing a pair of such derivatives, each containing a different glutamic acid linker between the NpG structure and the PSMA-targeting region, namely asymmetric urea. These compounds were named NpG-L-PSMA and NpG-D-PSMA.
First, the researchers ran tests using iodine-125 (125I) bound to these compounds rather than 211At, given that 125I is more abundant and easier to procure. Through experiments in mice bearing tumors from a human prostate cancer cell line, they found that both [125I]I-NpG-D-PSMA and [125I]I-NpG-L-PSMA exhibited low accumulation in the stomach and thyroid, hinting at their high in vivo stability against deiodination. However, [125I]I-NpG-D-PSMA showed higher accumulation in tumor tissue than [125I]I-NpG-L-PSMA.
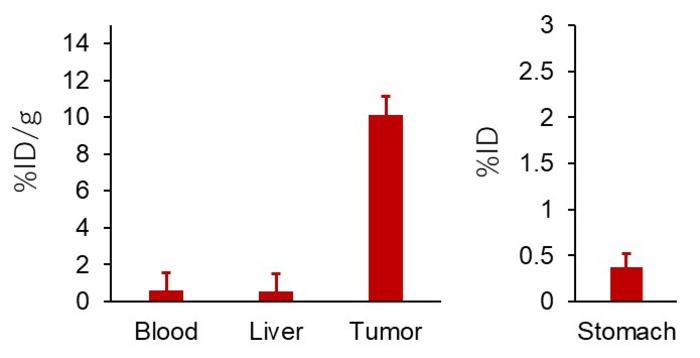
Thus, the team proceeded to run another series of experiments, now using [211At]At-NpG-D-PSMA. Just like its iodine-containing counterpart, this compound exhibited high accumulation in tumors and low accumulation in vital organs such as the liver and stomach.
Taken together, the results of these in vivo experiments highlight the potential of NpG-D-PSMA for targeted alpha therapy. “Our study showed that the neopentyl glycol structure, which can stably hold radiohalogens like 211At and 125I in vivo, may be applicable as a tumor-targeting agent. The use of the neopentyl glycol structure as a radiohalogen labeling moiety could enable the production of nuclear medicines for various types of tumors, thereby contributing greatly to human welfare,” concludes Uehara.
With any luck, further developments in this field will soon extend our arsenal against challenging metastatic cancers, lighting a beacon of hope for those affected.
About Professor Tomoya Uehara
Tomoya Uehara is a Principal Investigator and Professor at the Graduate School of Pharmaceutical Sciences, Chiba University. He specializes in the study and development of radiopharmaceuticals, both for diagnostic and therapeutic purposes, using techniques from analytical and physical chemistry. He has published over 80 peer-reviewed papers on these topics, and he is a member of the Japan Society of Nuclear Medicine, The Pharmaceutical Society of Japan, and the Japan Society for Molecular Imaging.
Journal
EJNMMI Radiopharmacy and Chemistry
Method of Research
Experimental study
Subject of Research
Animals
Article Title
In vivo stable 211At-labeled prostate-specific membrane antigen-targeted tracer using a neopentyl glycol structure
Article Publication Date
17-Jul-2024
COI Statement
No potential conflicts of interest relevant to this article exist.