In a groundbreaking advance poised to transform computational materials science, researchers at Harvard’s John A. Paulson School of Engineering and Applied Sciences (SEAS) have unveiled a novel machine learning framework that models complex material responses to electric fields with quantum-level precision. This innovative approach surmounts the limitations of traditional quantum mechanical simulations, enabling accurate modeling of systems comprising up to a million atoms—far surpassing the previous computational cap of a few hundred atoms. The implications of this development reach deep into materials design and energy technology, setting the stage for accelerated discovery and engineering of advanced functional materials.
The challenge that has long constrained materials modeling is the prohibitive computational cost of density functional theory (DFT), a quantum mechanical method that underpins much of our understanding of atomic and molecular interactions. While DFT boasts exceptional accuracy, its application is restricted to relatively small atomic ensembles due to the exponential increase in complexity with system size. This bottleneck has impeded simulations that seek to capture realistic materials behaviors at scales relevant to practical devices and processes.
Machine learning has emerged as a promising avenue to extend the reach of quantum simulations, offering ways to learn and interpolate quantum properties from limited datasets. However, existing machine learning models have struggled to accurately predict material responses to external perturbations such as electric fields. These challenges arise because many models neglect fundamental physical laws such as symmetry constraints and energy conservation tied to electrical polarization, resulting in predictions that can be inconsistent or physically implausible.
The team, led by Stefano Falletta along with Professor Boris Kozinsky, introduced a revolutionary method that embeds the essential physics directly into the learning architecture. Their approach, termed Allegro-pol, extends a previously established neural network model called Allegro. Whereas Allegro effectively captured atomic energies and forces, Allegro-pol unifies descriptions of energy, polarization, and electric response into a single, differentiable potential energy function that inherently respects quantum symmetries and conservation laws.
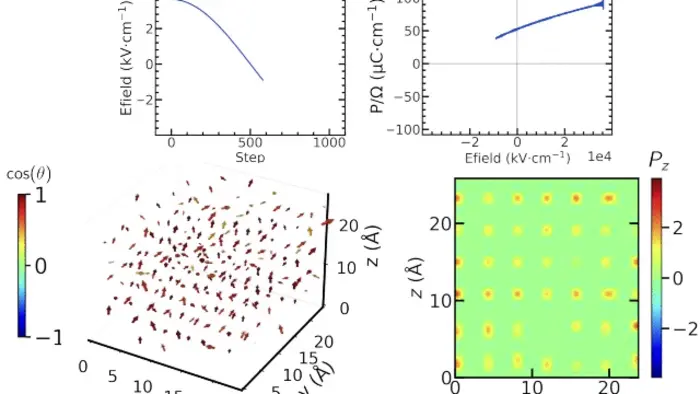
This framework is trained and rigorously validated against DFT computational data, allowing it to simulate how materials dynamically evolve under applied electric fields while ensuring that fundamental physics is upheld. Crucially, Allegro-pol captures the interplay between atomic structure and electric polarization, a key factor governing the behavior of ferroelectric and dielectric materials—categories of materials essential for non-volatile memory, capacitors, and energy storage technologies.
Beyond mere scalability, this work signifies a profound conceptual leap. By enabling simulations at scales on the order of hundreds of thousands to a million atoms with quantum mechanical fidelity, researchers can now explore mesoscale phenomena inaccessible to standard approaches. For instance, the team demonstrated the potential of Allegro-pol by simulating intricate infrared and electrical characteristics of silicon dioxide and modeling temperature-dependent ferroelectric switching in tetragonal barium titanate, a widely studied ferroelectric material. These applications validate the model’s accuracy in capturing polarization dynamics critical to device functionality.
This advance is not simply about accelerating computation but about integrating physical rigor with machine learning flexibility. Traditional black-box models fall short when confronted with complex quantum phenomena, but Allegro-pol’s architecture ensures internal consistency by embedding differentiable physics principles. This enables researchers to observe emergent behaviors in materials subject to external fields with unprecedented precision.
The implications extend far beyond the immediate applications demonstrated. Foundational models like Allegro-pol chart a new course for computational materials science, marrying the best of physics-based theory with data-driven methods. Such hybrid techniques promise to lower barriers in discovering novel materials with tailored electromagnetic, chemical, and mechanical properties, promising innovations in electronics, renewable energy, and more.
Falletta, now at Radical AI, underscored how this initiative is part of a broader trajectory towards integrating better theoretical models, expansive machine learning architectures, and advanced computational infrastructure. As clusters grow larger and GPU resources multiply, the ability to tackle increasingly complex materials problems in silico will transform scientific discovery pipelines.
The research community is particularly energized by the model’s potential to accurately simulate electrically driven phenomena, a realm that has traditionally resisted comprehensive modeling due to the interplay between electronic, ionic, and structural degrees of freedom. Allegro-pol’s capability to synthesize these elements into a coherent predictive framework opens new avenues not only for understanding fundamental materials physics but also for engineering practical devices.
This landmark study demonstrates the power of interdisciplinary collaboration—bringing together materials science, applied physics, computational chemistry, and machine learning—to solve long-standing computational bottlenecks. Through their work published in Nature Communications, the researchers provide a compelling example of how differentiable physics-informed machine learning can push the frontiers of scale and accuracy in materials modeling.
In essence, Allegro-pol represents a milestone in simulating electric response at scales and accuracy levels previously unimaginable. By seamlessly integrating quantum accuracy with machine learning scalability and physical constraints, this tool empowers scientists to probe and predict complex materials behavior under realistic operational conditions. The reverberations of this progress will be felt across fields where the electrical properties of materials are paramount, enabling the design of next-generation ferroelectric memories, capacitors, and energy storage systems that underpin our future technologies.
Subject of Research: Not applicable
Article Title: Unified differentiable learning of electric response
News Publication Date: 29-Apr-2025
Web References:
https://www.nature.com/articles/s41467-025-59304-1
References:
Falletta, S., Cepellotti, A., Johansson, A., Tan, C. W., Descoteaux, M. L., Musaelian, A., Owen, C. J., Kozinsky, B. (2025). Unified differentiable learning of electric response. Nature Communications. https://doi.org/10.1038/s41467-025-59304-1
Image Credits: Stefano Falletta
Keywords: Artificial intelligence, Computer science, Applied mathematics, Equations, Mathematical analysis, Chemistry, Chemical modeling, Materials science, Material properties, Chemical properties, Electromagnetic properties, Physics, Computational physics, Computational chemistry, Quantum mechanics, Density functional theory, Quantum dynamics

{kind=link}