{kind=link}
Unlocking biological information from complex single-cell genomic data has just become easier and more precise, thanks to the innovative ‘scLENS’ tool developed by the Biomedical Mathematics Group within the IBS Center for Mathematical and Computational Sciences led by Chief Investigator KIM Jae Kyoung, who is also a Professor at KAIST. This new finding represents a significant leap forward in the field of single-cell transcriptomics.

Credit: Institute for Basic Science
Unlocking biological information from complex single-cell genomic data has just become easier and more precise, thanks to the innovative ‘scLENS’ tool developed by the Biomedical Mathematics Group within the IBS Center for Mathematical and Computational Sciences led by Chief Investigator KIM Jae Kyoung, who is also a Professor at KAIST. This new finding represents a significant leap forward in the field of single-cell transcriptomics.
Single-cell genomic analysis is an advanced technique that measures gene expression at the individual cell level, revealing cellular changes and interactions that are not observable with traditional genomic analysis methods. When applied to cancer tissues, this analysis can delineate the composition of diverse cell types within a tumor, providing insights into how cancer progresses and identifying key genes involved during each stage of progression.
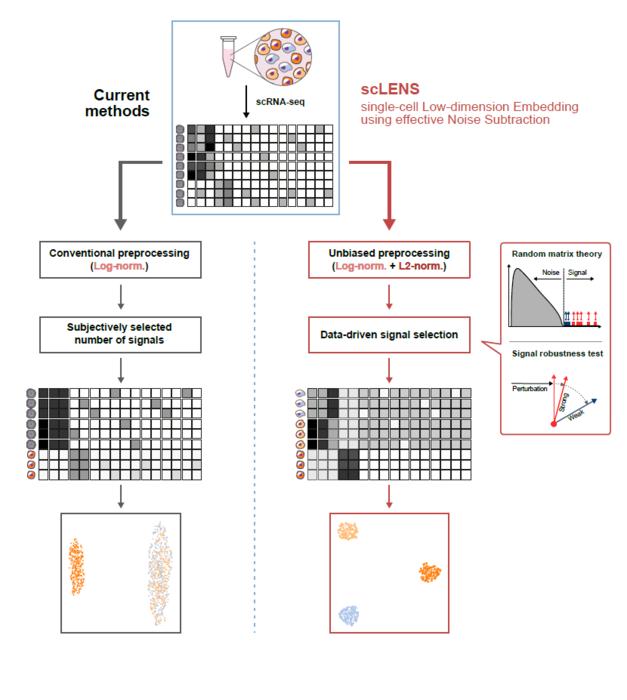
Despite the immense potential of single-cell genomic analysis, handling the vast amount of data that it generates has always been challenging. The amount of data covers the expression of tens of thousands of genes across hundreds to thousands of individual cells. This not only results in large datasets but also introduces noise-related distortions, which arise in part due to current measurement limitations.
Corresponding author KIM Jae Kyoung highlighted, “There has been a remarkable advancement in experimental technologies for analyzing single-cell transcriptomes over the past decade. However, due to limitations in data analysis methods, there has been a struggle to fully utilize valuable data obtained through extensive cost and time.”
Researchers have developed numerous analysis methods over the years to discern biological signals from this noise. However, the accuracy of these methods has been less than satisfactory. A critical issue is that determining signal and noise thresholds often depends on subjective decisions from the users.
The newly developed scLENS tool harnesses Random Matrix Theory and Signal robustness test to automatically differentiate signals from noise without relying on subjective user input.
First author KIM Hyun stated, “Previously, users had to arbitrarily decide the threshold for signal and noise, which compromised the reproducibility of analysis results and introduced subjectivity. scLENS eliminates this problem by automatically detecting signals using only the inherent structure of the data.”
During the development of scLENS, researchers identified the fundamental reasons for inaccuracies in existing analysis methods. They found that commonly used data preprocessing methods distort both biological signals and noise. The new preprocessing approach that scLENS offers is free from such distortions.
By resolving issues related to noise threshold determined by subjective user choice and signal distortion in conventional data preprocessing, scLENS significantly outperforms existing methods in accuracy. Additionally, scLENS automates the laborious process of signal dimension selection, allowing researchers to extract biological signals conveniently and automatically.
CI Kim added, “scLENS solves major issues in single-cell transcriptome data analysis, substantially improving the accuracy and efficiency throughout the analysis process. This is a prime example of how fundamental mathematical theories can drive innovation in life sciences research, allowing researchers to more quickly and accurately answer biological questions and uncover secrets of life that were previously hidden.”
This research was published in the international journal ‘Nature Communications’ on April 27.
Terminology
* Single-cell RNA sequencing (scRNA-seq): A technique used to measure gene expression levels in individual cells, providing insights into cell heterogeneity and rare cell types.
* Dimensionality reduction: A method to reduce the number of features or variables in a dataset while preserving the most important information, making data analysis more manageable and interpretable.
* Random matrix theory: A mathematical framework used to model and analyze the properties of large, random matrices, which can be applied to filter out noise in high-dimensional data.
* Signal robustness test: Among the signals, this test selects signals that are robust to the slight perturbation in data because real biological signals should be invariant for such slight modification in the data.
Journal
Nature Communications
Method of Research
Computational simulation/modeling
Subject of Research
Cells
Article Title
scLENS: Data-driven signal detection for unbiased scRNA-seq data analysis
Article Publication Date
27-Apr-2024