A pioneering study led by Professor ZHAO Guoping and his research team at the Hefei Institutes of Physical Science, Chinese Academy of Sciences, has uncovered an unprecedented molecular mechanism that governs DNA damage repair, with profound implications for cancer therapy and radiotherapy sensitivity. The research draws back the curtain on the complex orchestration of post-translational modifications that regulate the fate and functionality of key DNA repair proteins. Published in the prestigious journal Cell Death & Differentiation, this study advances our understanding of how tumor cells leverage such repair pathways to evade the lethal effects of ionizing radiation, a frontline therapeutic strategy for numerous cancers.
Radiotherapy fundamentally operates by inflicting critical DNA lesions, predominantly double-strand breaks (DSBs), in target cancer cells. These lesions are inherently cytotoxic, and their efficient repair directly correlates with tumor cell survival post-treatment. However, many tumors, such as breast and lung cancers, express abnormally elevated levels of repair proteins, which enable brisk and robust repair of DNA damage, culminating in treatment resistance. The intricate network underlying this repair process is heavily influenced by epigenetic and post-translational modifications, realms that have remained partially opaque until now. Prof. ZHAO’s team delved deeply into this regulatory labyrinth, focusing on the role of SUMOylation, a small ubiquitin-like modifier process, in modulating DNA repair dynamics.
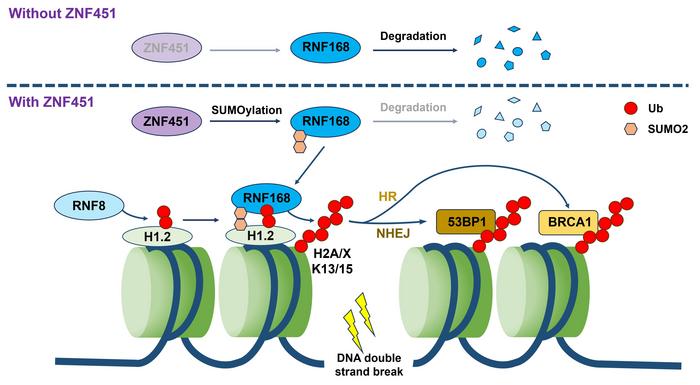
Central to their discovery is the zinc finger protein ZNF451, which the investigators identified as markedly overexpressed in multiple malignancies, including breast and lung cancers. Importantly, elevated ZNF451 levels were correlated with poorer prognostic outcomes in patient cohorts, signaling its potential clinical relevance. Upon exposure to ionizing radiation, ZNF451 rapidly localizes to DNA damage sites, initiating a catalytic event that post-translationally modifies the ubiquitin ligase RNF168 via conjugation with the SUMO2 modifier. This SUMOylation event acts as a stabilization mechanism for RNF168, preventing its proteasomal degradation and thereby augmenting its accumulation and retention at the sites of DNA lesions.
RNF168 itself is a pivotal E3 ubiquitin ligase responsible for propagating ubiquitin signaling cascades on chromatin, particularly on histone variants such as H2A and H2AX. These histone ubiquitinations are essential for recruiting downstream effector proteins that execute DNA repair through homologous recombination or non-homologous end joining. By enhancing RNF168 stability and localization, ZNF451 effectively amplifies the ubiquitin signal at damaged chromatin regions, thereby potentiating the cellular DNA repair response. This amplification mechanism is especially critical in the context of radiotherapy, where DNA damage must be resolved swiftly to facilitate tumor cell survival.
Intriguingly, the research also unveiled a sophisticated crosstalk between ZNF451 and RNF8, another canonical E3 ligase involved early in the DNA damage response cascade. Contrary to straightforward cooperative behavior, these two proteins exhibit a dynamic regulatory network characterized by competitive binding to RNF168, effectively regulating its recruitment and activity in a finely balanced manner. The study’s quantitative analyses revealed that ZNF451 and RNF8 exert mutually inhibitory influences on each other’s ability to recruit RNF168, yet paradoxically, simultaneous depletion of both proteins caused a severe deficiency in RNF168 accumulation at DNA damage sites. This underscores a complex equilibrium that maintains optimal ubiquitination signaling necessary for efficient DNA repair.
The sum of these findings led the authors to propose a novel model of “dynamic equilibrium regulation” in DNA damage repair, wherein opposing but complementary interactions between different ligases tune the amplitude and duration of ubiquitin signaling. Such a model has profound implications for understanding how tumor cells orchestrate DNA repair and resist genotoxic therapies. The elucidation of this balance between SUMOylation and ubiquitination at the molecular level opens potential avenues for therapeutic intervention by disrupting this equilibrium to sensitize tumors to radiation-induced damage.
This breakthrough also emphasizes the emerging importance of SUMOylation in the DNA damage response. While ubiquitination has been extensively studied in this context, SUMO modifications add an additional layer of regulation, influencing the stability, localization, and interactions of key repair factors. SUMO2 conjugation of RNF168 represents one such modification that stabilizes this integral repair protein, illustrating the nuanced interplay between different post-translational modifications in fine-tuning repair pathways.
The work by ZHAO et al. not only delineates the mechanistic insights into SUMO-mediated regulation of DNA repair but also highlights the translational potential of targeting the ZNF451-RNF8-RNF168 axis. Inhibiting the SUMOylation activity of ZNF451 or manipulating its interaction with RNF8 could render tumor cells more susceptible to radiotherapy, overcoming intrinsic radioresistance. This approach could revolutionize treatment paradigms for cancers characterized by DNA repair deregulation.
Furthermore, the identification of ZNF451’s overexpression as a biomarker linked to poor prognosis offers a clinical window for patient stratification. Patients with high ZNF451 expression may require more aggressive or alternative therapeutic regimes to counterbalance their tumors’ enhanced DNA repair capacity. This personalized medicine angle adds another valuable dimension to the research’s clinical impact.
In summary, this research unveiled a previously underappreciated regulatory axis centered on ZNF451’s SUMOylation of RNF168, modulated by the interplay with RNF8, that collectively amplifies ubiquitin signaling essential for DNA repair. This intricate regulatory schema redefines our comprehension of how cells dynamically control repair factor activities at DNA lesions and how disruptions in this system influence cancer radiotherapy sensitivity. As the field moves forward, targeting this finely tuned equilibrium holds promise as a novel therapeutic strategy to enhance the efficacy of DNA-damaging treatments.
This landmark study, set to transform the landscape of cancer biology and therapy, eloquently demonstrates the power of dissecting post-translational modification networks in revealing vulnerabilities within tumor DNA repair machinery. Through meticulous experimental work and mechanistic elucidation, Prof. ZHAO’s team has not only expanded the molecular vocabulary of DNA repair regulation but has also paved the way toward novel radiosensitization approaches critical for improving patient outcomes in oncology.
Subject of Research: Regulation of DNA damage repair mechanisms via SUMOylation and ubiquitination, focusing on the role of ZNF451, RNF8, and RNF168 in modulating radiotherapy sensitivity in cancer.
Article Title: ZNF451 collaborates with RNF8 to regulate RNF168 localization and amplify ubiquitination signaling to promote DNA damage repair and regulate radiosensitivity
News Publication Date: 7-Mar-2025
Web References: http://dx.doi.org/10.1038/s41418-025-01472-0
Image Credits: ZHAO Guoping
Keywords: Physical sciences

{kind=link}