In the intricate world of cellular biology, mitochondria have long been recognized as the powerhouses that fuel the cell’s energy demands. These semi-autonomous organelles harbor their own distinct DNA, known as mitochondrial DNA (mtDNA), which is separate from the nuclear genome. Notably, mtDNA exhibits a mutation rate significantly higher than that of nuclear DNA, a phenomenon that has been observed with increasing frequency in aging tissues as well as in various malignancies. Despite these observations, the direct involvement of mtDNA mutations in the onset and progression of cancer has remained elusive and controversial within the scientific community.
A groundbreaking study spearheaded by Dr. Zhenglong Gu, Director of the Center for Mitochondrial Genetics and Health at Fudan University and Courtesy Professor at Cornell University, offers compelling new evidence that positions heteroplasmic mutations in the mitochondrial gene MT-ND5 as critical drivers of cancer initiation. Published in the journal Mitochondrial Communications, this research delves into the molecular underpinnings by which mutations in MT-ND5, a gene encoding an essential subunit of mitochondrial complex I, disrupt oxidative phosphorylation and propel oncogenic transformation.
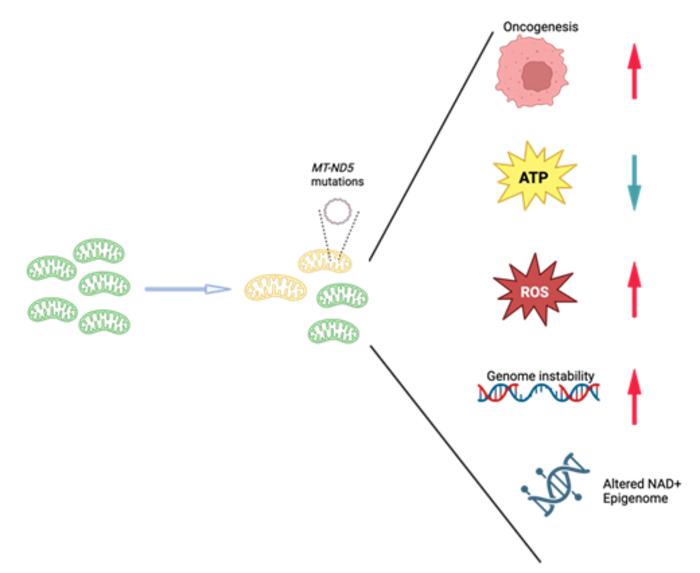
The study employed rigorous experimental methodologies to introduce de novo mutations into the MT-ND5 gene, thereby creating cellular models that mimic heteroplasmy—a condition where mutant and wild-type mtDNA coexist within the same cell. This nuanced approach enabled the investigators to systematically dissect how varying levels of heteroplasmy affect mitochondrial function. Their findings revealed that even low to moderate heteroplasmic burdens of MT-ND5 mutations are sufficient to impair complex I activity, leading to a marked increase in mitochondrial reactive oxygen species (ROS). This oxidative stress, in turn, appears to heighten the cells’ oncogenic potential significantly.
Intriguingly, the metabolic rewiring associated with MT-ND5 heteroplasmy was characterized by a pronounced shift from oxidative phosphorylation to glycolysis, aligning with the well-known Warburg effect observed in cancer cells. However, Dr. Gu’s team uncovered an unexpected twist to this metabolic adaptation: the shift toward glycolysis serves not primarily to meet energetic demands but rather to restore NAD⁺ pools, which are essential cofactors in numerous metabolic and signaling pathways. Measurements demonstrated that despite partial reductions in mutant mtDNA levels, NAD⁺ concentrations failed to recover fully, underscoring a persistent metabolic vulnerability linked to altered mitochondrial genetics.
Beyond establishing a causal link between specific mtDNA mutations and cancer initiation, this study extended its scope to investigate cellular quality control mechanisms governing the retention and tolerance of deleterious mtDNA variants. Longitudinal tracking of mitochondrial heteroplasmy and phenotypic manifestations illuminated complex regulatory pathways that maintain mitochondrial genome integrity over time, even in the face of mutational insults. These insights suggest that cells balance the functional costs of harboring mutant mitochondria against the need to preserve energy homeostasis and genomic stability.
The implications of these findings are profound, signaling a paradigm shift in our understanding of oncogenesis. Whereas previous frameworks predominantly emphasized nuclear genomic alterations as the primary culprits, Dr. Gu’s work spotlights mitochondrial genetic dynamics as indispensable contributors to cancer biology. This dual-genome perspective opens new avenues for precision medicine strategies aimed at early detection, prevention, and targeted therapy of cancers rooted in mitochondrial dysfunction.
Despite these advances, the research team acknowledges significant gaps remain in deciphering the complex interplay between mtDNA mutations and the nuclear genomic environment within tumorigenic contexts. Future investigations are poised to explore how multiple mtDNA variants interact with diverse nuclear backgrounds to modulate cancer risk and progression. Such integrative studies will be pivotal in unraveling the multifaceted genetic networks that orchestrate cellular transformation.
Moreover, the study’s methodological approach—leveraging both in vitro cell culture and in vivo animal models—provides robust validation of the oncogenic capacity conferred by MT-ND5 heteroplasmy. This dual-platform analysis strengthens the translational potential of the findings and lays the groundwork for therapeutic exploration targeting mitochondrial genome maintenance and metabolic reprogramming in oncology.
In conclusion, the elucidation of mitochondrial heteroplasmic mutations as active architects of oncogenesis represents a milestone in molecular biology and cancer research. By illuminating the nuanced relationship between mtDNA integrity, metabolic adaptation, and cellular transformation, Dr. Gu and his collaborators have paved the way for innovative precision medicine paradigms that account for mitochondrial genetics in cancer risk assessment and intervention.
As Dr. Gu articulates, "Our research highlights the often-overlooked mitochondrial genome’s role in the complex landscape of cancer initiation. Understanding how heteroplasmic MT-ND5 mutations drive tumorigenesis not only advances fundamental science but also charts a promising path toward personalized cancer prevention and prediction."
This pioneering research reinforces the necessity of expanding the genetic and metabolic lens through which cancer is studied and treated, underscoring mitochondria as both energetic and genetic arbiters of cellular fate.
Subject of Research: Cells
Article Title: [Not provided]
News Publication Date: [Not provided]
Web References: http://dx.doi.org/10.1016/j.mitoco.2025.03.001
References: Gu Z, et al. Mitochondrial Communications, 2025.
Image Credits: Yuanyuan et al.
Keywords: Life sciences, Molecular biology, Cancer, Nutrients, Nutrition

{kind=link}