A remarkable breakthrough in cardiovascular disease research has emerged from the latest study by Du, Wan, Du, and colleagues, published in Nature Communications. Their investigation unveils a novel molecular mechanism by which the enzyme MAT2A orchestrates the destabilization of atherosclerotic plaques, a leading contributor to heart attacks and strokes. This groundbreaking study provides compelling insight into how epigenetic reprogramming of macrophages—immune cells central to atherosclerosis progression—is mediated through MAT2A, offering promising new avenues for therapeutic intervention.
Atherosclerosis, a chronic inflammatory condition characterized by the buildup of fatty plaques within arterial walls, poses a persistent threat to global health. While numerous studies have explored lipid metabolism and immune responses within plaques, the precise molecular drivers of plaque vulnerability—the propensity to rupture and cause acute cardiovascular events—remain incompletely understood. Here, the multidisciplinary team focused on dissecting how epigenetic modifications in macrophages influence plaque stability, revealing a pivotal role for methionine adenosyltransferase 2A (MAT2A).
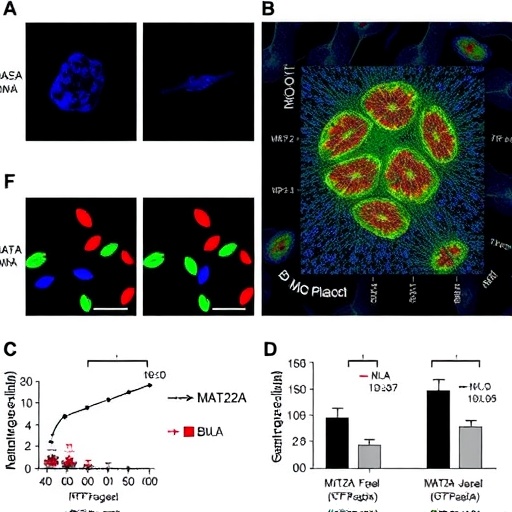
MAT2A, an enzyme traditionally known for its role in catalyzing the synthesis of S-adenosylmethionine (SAM), a universal methyl donor, was identified as a key modulator of macrophage function within atherosclerotic environments. The researchers demonstrated that elevated MAT2A activity promotes a reprogramming of macrophage epigenomes, specifically reshaping histone methylation landscapes. These epigenetic alterations result in a transcriptional shift that favors pro-inflammatory and matrix-degrading phenotypes, thereby compromising plaque structural integrity.
Utilizing a combination of in vivo mouse models and ex vivo human plaque samples, the study provided rigorous evidence linking MAT2A upregulation to plaque vulnerability markers. Advanced chromatin immunoprecipitation sequencing (ChIP-seq) and RNA-seq analyses delineated the genome-wide shifts in histone methylation and gene expression patterns orchestrated by MAT2A. Particularly notable was the enrichment of H3K27 trimethylation changes at loci controlling extracellular matrix remodeling and inflammatory cytokine production, implicating these pathways in the pathological remodeling of plaques.
Further mechanistic interrogation revealed that pharmacological inhibition or genetic silencing of MAT2A curbed the pro-inflammatory macrophage phenotype and fortified plaque architecture. Treated animals exhibited reduced rates of plaque rupture and a lower incidence of downstream ischemic events. These findings underscore MAT2A’s potential as a therapeutic target, highlighting the feasibility of epigenetic modulation to stabilize plaques and prevent catastrophic cardiovascular outcomes.
The implications of this study are profound. By linking one of the core metabolic enzymes responsible for methyl group donation to epigenetic reprogramming within lesions, the research expands the conceptual framework of atherosclerosis pathogenesis. It elucidates how metabolic-epigenetic crosstalk within immune cells drives disease progression, integrating molecular biology, immunology, and vascular medicine in an unprecedented manner. Such insights pave the way for developing next-generation therapies that transcend lipid lowering and inflammation control by modulating chromatin state directly.
Moreover, the study’s methodological rigor and translational relevance set a new standard for cardiovascular epigenomics research. The utilization of cutting-edge multi-omics techniques allowed for a granular dissection of the MAT2A-dependent methylome changes, while in vivo functional assays validated their pathogenic significance. This comprehensive analytical approach not only delineates causative molecular events but also affirms the potential of epigenetic enzyme inhibitors as drug candidates.
Beyond its immediate impact on atherosclerosis research, this discovery has broader ramifications for other inflammation-related chronic diseases where macrophage plasticity plays a determinant role. Since epigenetic regulation governs immune cell phenotype switching, interventions targeting enzymes like MAT2A could modulate immune responses across diverse pathological contexts, making this a versatile therapeutic strategy with wide-ranging applications.
This pioneering investigation also highlights the intricate relationship between metabolism and epigenetics in shaping immune cell functions. Given that SAM synthesis via MAT2A directly controls methylation capacities, the enzyme serves as a metabolic-epigenetic nexus. This dual role could underlie how environmental factors such as diet influence epigenetic landscapes in immune cells, further impacting cardiovascular risk profiles—a fertile area for future research.
Intriguingly, the study identifies MAT2A as a node amenable to pharmacological manipulation without broadly suppressing systemic immune functions. This specificity minimizes the likelihood of adverse effects typically associated with immunosuppressive therapies, offering a safer therapeutic window. The development of selective MAT2A inhibitors, therefore, emerges as a tangible goal to mitigate plaque vulnerability while preserving immune homeostasis.
Furthermore, the research underscores the importance of macrophage heterogeneity within plaques, revealing distinct epigenetic states that govern functional fate decisions. These findings call for a refinement of current macrophage classification schemes based on epigenomic and transcriptomic signatures, enhancing our understanding of cellular dynamics in diseased vessels.
As cardiovascular disease remains the leading cause of mortality worldwide, innovations in pinpointing molecular determinants of plaque destabilization are urgently needed. The demonstration that epigenetic reprogramming via MAT2A is instrumental in plaque vulnerability constitutes a significant leap forward in this quest. Translating these insights into clinical practice could revolutionize patient risk stratification and therapeutic management.
In summary, Du et al.’s study represents a milestone in cardiovascular biology, illuminating the fundamental mechanisms by which epigenetic modulation of macrophages drives atherosclerotic plaque instability. Their work not only advances scientific knowledge but also charts a new course toward precision medicine strategies targeting chromatin-modifying enzymes. This paradigm shift holds promise for reducing the global burden of cardiovascular events through innovative epigenetic therapies.
As researchers and clinicians integrate these findings into ongoing studies and therapeutic designs, the potential to mitigate heart attack and stroke risk by harnessing epigenetic regulation emerges as a compelling frontier. Continued exploration of MAT2A’s role, alongside other metabolic-epigenetic interconnections, will undoubtedly accelerate the development of next-generation cardiovascular interventions poised to save millions of lives.
Subject of Research: Atherosclerotic plaque vulnerability and epigenetic reprogramming of macrophages mediated by MAT2A.
Article Title: MAT2A promotes atherosclerotic plaque vulnerability by mediating epigenetic reprogramming of macrophages.
Article References:
Du, Z., Wan, P., Du, M. et al. MAT2A promotes atherosclerotic plaque vulnerability by mediating epigenetic reprogramming of macrophages. Nat Commun 16, 11168 (2025). https://doi.org/10.1038/s41467-025-66121-z
Image Credits: AI Generated
DOI: https://doi.org/10.1038/s41467-025-66121-z

{kind=link}