{kind=link}
In a paper recently published in Nature Communications, the HUN-REN-ELTE Protein Modeling Research Group (Institute of Chemistry) has laid the foundations for a mathematical method, allowing the computer-assisted comparison of the three-dimensional structures of proteins. The method is unique in that while the alternatives available so far only took into account the position of the atoms, the new technique, called LoCoHD (Local Composition Hellinger Distance), also includes the chemical information of the atoms.

Credit: Image: Eötvös Loránd University
In a paper recently published in Nature Communications, the HUN-REN-ELTE Protein Modeling Research Group (Institute of Chemistry) has laid the foundations for a mathematical method, allowing the computer-assisted comparison of the three-dimensional structures of proteins. The method is unique in that while the alternatives available so far only took into account the position of the atoms, the new technique, called LoCoHD (Local Composition Hellinger Distance), also includes the chemical information of the atoms.
Proteins are molecular machines that carry out processes necessary for cells to function, acting as molecular switches, transcribing information from DNA, transporting small and large molecules and regulating metabolism related chemical reactions. However, for all this to succeed, the protein in question must have the right spatial conformation, i.e. its own, correct 3D arrangement. Several experimental methods (X-ray crystallography, nuclear magnetic resonance spectroscopy, cryo-electron microscopy) are available to determine the arrangement of atoms in a protein, and over the last few decades protein researchers have discovered the shape of nearly 220,000 proteins. These results increasingly demand the development of computational methods capable of analyzing these arrangements.
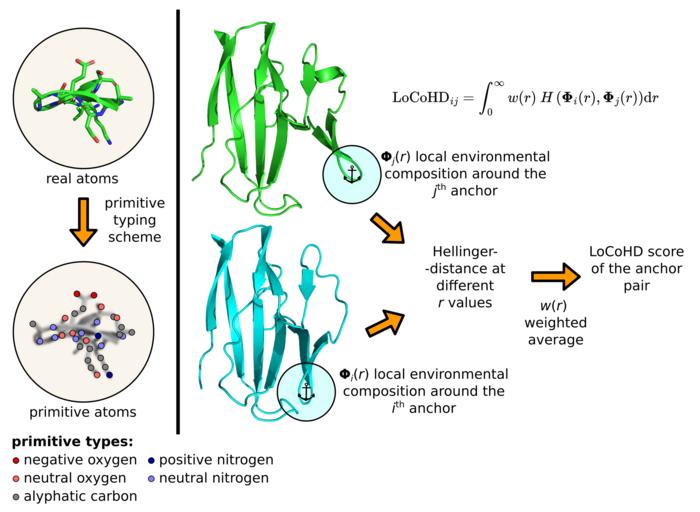
One such method is the algorithm called LoCoHD, developed by Zsolt Fazekas, a PhD candidate at the ELTE Hevesy György School of Chemistry and a researcher in Dr. András Perczel’s research group. The algorithm compares local environments around amino acids in proteins based on their chemical nature (e.g. elemental composition, charge, hydrophobicity, etc.). The method decides on a simple scale of 0 to 1 how different the structures in question are from each other. Values close to 0 suggest a high similarity between atomic arrangements and chemical properties, while values close to 1 indicate that the proteins being compared may have very different properties. The resulting numerical value (a so-called metric) can thus be used to obtain new information about the system under study.
The algorithm uses a multi-step protocol to generate the number representing the structural differences. In the first step, it converts real atoms in the protein into so-called primitive atoms. These can be represented as virtually labeled positionso whose labels tell the chemical nature of the original atom. So, for example, an primitive atom can be a “positively charged nitrogen”, a “negatively charged oxygen”, a “neutrally charged oxygen”, an “aromatic carbon”, etc. The labels are generated according to a so-called primitive typing scheme, which tells us in a tabulated manner how to convert real atoms into primitive atoms. The user can freely specify this table, fixing the chemical resolution of the method. The second step is to determine the reference points of the comparison by selecting a subset of primitive atoms. These selected special primitive atoms are called the anchor atoms. For each selected anchor atom pair, the algorithm performs a comparison step, the result of which gives the dissimilarity measure we want. These numbers can be used at a local level, or they can be averaged into a single descriptor characterizing the whole protein.
In the study, published in the prestigious journal Nature Communications, the researchers highlighted that the method can also be used in the biannual CASP (Critical Assessment of Protein Structure Prediction) competitions, which is a well known competition in the field of protein research. During this event, competitors use different algorithms to model the shape of proteins having yet unpublished structures. CASP judges use a number of structure comparison methods to evaluate the contenders, but none of these take into account the chemistry of the local amino acid environments. Using data from the 2020 CASP14 competition, the researchers have now performed comparative analysis of several modeled proteins, including the structures predicted by the artificial-intelligence-based AlphaFold2 method. Among these, they highlighted the analysis of a protein from the SARS-CoV-2 virus called ORF8. In the modeled structures of this protein, amino acid environments were identified that differ significantly in their interaction patterns from the environments found in the experimental structure.
In addition to studying static structures, the researchers also tested whether the method is suitable for analyzing the internal motion of proteins. They used simulations capable of reproducing molecular motions and data extracted from structural ensembles. One of the systems under study was the podocin protein, which performs vital functions in the kidney and whose mutations can cause severe, often fatal conditions. The LoCoHD method was used to identify amino acids in the protein that undergo major chemical-environmental changes during the movement of podocin, which can affect both its structure and function. Similarly, the LoCoHD method has been applied successfully in the study of the HIV-1 capsid protein, in which an amino acid critical for the formation of the viral envelope has been identified.
These results are not only research curiosities, but by studying protein structures more effectively, we can get closer to better understanding the pathogens causing severe diseases and to developing effective drugs and therapeutics.
Journal
Nature Communications
Article Title
LoCoHD: a metric for comparing local environments of proteins
Article Publication Date
13-May-2024