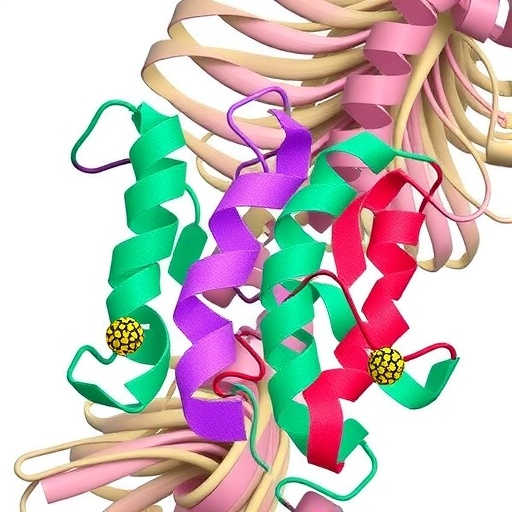
In the relentless quest to unravel the molecular underpinnings of hypertrophic cardiomyopathy (HCM), a genetic disorder that often leads to sudden cardiac death, a groundbreaking study has emerged that sheds new light on the intricate allosteric mechanisms of human β-cardiac myosin. Published in Nature Communications in 2025, the research meticulously investigates how two specific mutations—Y115H and E497D—subvert the biomechanical function of myosin, a motor protein essential for cardiac muscle contraction. These mutations are shown not merely to alter myosin’s active site but to disrupt a critical conformational state known as the folded-back state, thereby offering a refined molecular perspective on HCM pathology.
β-cardiac myosin is a powerhouse protein underpinning the contractile function of heart muscle cells. It functions by undergoing a series of conformational changes culminating in the generation of force and movement along actin filaments. A key aspect of its regulation involves the folded-back state, a structurally compact autoinhibited conformation that ensures myosin’s activity is tightly controlled to prevent excessive energy consumption and maintain cardiac efficiency. The folded-back state, sometimes referred to as the interacting-heads motif, reduces myosin’s ATPase activity, acting as a physiological brake during periods when diminished contraction is needed.
The study led by Nandwani, Bhowmik, Glaser, and colleagues uses sophisticated biochemical and biophysical approaches, including cryo-electron microscopy and molecular dynamics simulations, to reveal how mutations Y115H and E497D, located remotely from the nucleotide-binding site, exert allosteric effects that unravel this folded-back state. These mutations lie in regions of the myosin molecule that are critical for stabilizing the autoinhibited conformation, and their perturbation shifts the equilibrium away from this energy-conserving form.
By destabilizing the folded-back state, the mutations effectively unleash a hypercontractile phenotype at the molecular level. This aberrant conformational unlocking enhances myosin head availability and increases ATPase activity, leading to excessive force generation and dysregulated cardiac muscle contraction. Such molecular hyperactivity manifests clinically as hypertrophic cardiomyopathy, characterized by thickened cardiac walls and impaired relaxation, ultimately predisposing patients to arrhythmias and heart failure.
A remarkable aspect of the findings is the allosteric nature of the mutation effects. Unlike classical paradigms where mutations in enzymatic active sites directly influence catalysis, these HCM mutations operate via distant intramolecular communication pathways that can drastically alter protein dynamics and stability. This insight elevates understanding of protein allostery in muscle disease and underscores the potential of targeting allosteric sites for therapeutic intervention.
The authors provide compelling structural data delineating how Y115H and E497D mutations modify the interactions among the myosin heads and their associated tail domains. Cryo-EM reconstructions capture the conformational shifts at near-atomic resolution, unveiling how side chain substitutions reverberate through the protein scaffold to hinder formation of the folded-back architecture. Molecular dynamics simulations complement these observations, offering dynamic perspectives on the destabilization processes in physiological ionic environments.
Functionally, the mutant myosins exhibit increased actin sliding velocity and heightened basal ATPase rates when compared to wild-type β-cardiac myosin, validating the structural premise of increased contractile activity. This enhanced activity correlates with clinical phenotypes of HCM, where excessive myosin function leads to maladaptive myocardial remodeling. Intriguingly, the study also hints at potential compensatory mechanisms that the heart might deploy to counterbalance this hypercontractility, a subject warranting further investigation.
Crucially, the elucidation of mechanistic pathways underlying these two specific mutations widens the spectrum of genotype-phenotype correlations in HCM. While previous research has focused predominantly on mutations within the motor domain, this study emphasizes the broader impact of allosteric regulatory regions, thus providing a more nuanced framework for genetic screening and risk stratification in affected individuals.
This work further propels the field toward rational drug design aimed at rescuing the folded-back state or mimicking its stabilizing effect. Targeting allosteric hotspots identified within this study could lead to novel pharmacological agents capable of normalizing myosin function without impeding its essential contractile properties. Such precision therapeutics could transform clinical management strategies, alleviating the burden of HCM worldwide.
In addition to clinical implications, the study’s integrated methodological approach exemplifies the power of combining advanced imaging with computational modeling to dissect complex protein dynamics. This blueprint may be extended to investigate other sarcomeric proteins implicated in cardiomyopathies, widening the horizons of molecular cardiology.
The pathogenesis of hypertrophic cardiomyopathy being deeply intertwined with subtle disruptions in sarcomeric protein function, the insights gleaned here showcase how even single amino acid alterations in remote regions of a protein can cascade into global conformational and functional disturbances. This highlights a broader biological principle: the delicate balance of protein folding and function is critically dependent on finely tuned intramolecular interactions that can be vulnerable to genetic perturbations.
Reflecting on the implications of this research, clinicians and researchers alike stand at a pivotal juncture. The capacity to delineate the precise molecular consequences of mutations opens the door to personalized medicine approaches where therapeutic strategies can be customized based on the mutation-driven conformational defects identified in individual patients.
Furthermore, the study addresses a pressing gap in our understanding of how energy efficiency and mechanical performance are coordinated at the molecular level in cardiac muscle. This coordination is vital to the heart’s ability to meet fluctuating physiological demands, and unraveling it at such a granular level provides foundational knowledge for tackling not only HCM but potentially other forms of heart failure.
The findings from Nandwani and colleagues’ team ultimately point to a unifying model of myosin regulation that integrates nucleotide binding, mechanical strain, and conformational states governed by allosteric networks. This integrative perspective enriches the conceptual landscape of muscle biology and paves the way for future discovery.
In sum, this seminal research represents a significant leap forward in molecular cardiology, combining cutting-edge technology with profound clinical insights. By clarifying how mutations Y115H and E497D disrupt the folded-back state of human β-cardiac myosin allosterically, the study propels the field closer to deciphering the molecular choreography that sustains healthy heart function, and to devising innovative solutions for hypertrophic cardiomyopathy—one of the most enigmatic and deadly cardiac diseases.
Subject of Research: The allosteric disruption of the folded-back state in human β-cardiac myosin by hypertrophic cardiomyopathy mutations Y115H and E497D.
Article Title: Hypertrophic cardiomyopathy mutations Y115H and E497D disrupt the folded-back state of human β-cardiac myosin allosterically.
Article References:
Nandwani, N., Bhowmik, D., Glaser, C. et al. Hypertrophic cardiomyopathy mutations Y115H and E497D disrupt the folded-back state of human β-cardiac myosin allosterically. Nat Commun 16, 8751 (2025). https://doi.org/10.1038/s41467-025-63816-1
Image Credits: AI Generated

{kind=link}