A recent groundbreaking study published in Nature Communications has unveiled a pivotal mechanism by which idiopathic pulmonary fibrosis (IPF) progresses, shining a spotlight on the enzyme histone deacetylase 11 (Hdac11). This discovery opens new avenues for therapeutic intervention in a disease that currently lacks effective treatment options and is marked by relentless lung scarring and respiratory decline. Researchers led by Nie, Xu, and Liu have identified Hdac11 as a key promoter of the M2 polarization state of macrophages—immune cells implicated in tissue remodeling and fibrosis. By curbing a critical mitochondrial quality control process known as Parkin-dependent mitophagy, Hdac11 fosters an environment conducive to myofibroblast accumulation, the very cells responsible for excessive extracellular matrix deposition in IPF.
Idiopathic pulmonary fibrosis is a devastating disorder characterized by the progressive thickening and stiffening of lung tissue through abnormal fibroblast activity, which obliterates the delicate alveolar architecture necessary for efficient gas exchange. Until now, the molecular underpinnings steering macrophage behavior and their communication with fibroblasts remained incomplete, thereby limiting targeted therapies. The current study meticulously dissects the crosstalk between epigenetic regulation and mitochondrial homeostasis in macrophages, revealing how Hdac11 suppression of the Parkin protein disrupts mitophagy pathways. This impairment fuels an inflammatory and fibrotic milieu through macrophage skewing toward a pro-fibrotic M2 phenotype.
Histone deacetylases like Hdac11 belong to an enzyme family pivotal in modulating chromatin structure and gene expression by removing acetyl groups from histone proteins, thereby tightening DNA packaging and repressing gene transcription. Unlike classical Hdacs more widely studied in cancer and neurological disorders, Hdac11 has remained relatively enigmatic until now. The researchers demonstrate how Hdac11 specifically downregulates Parkin, an E3 ubiquitin ligase central to selectively removing damaged mitochondria via mitophagy. The resulting mitochondrial dysfunction promotes macrophage alternative activation, a state linked to tissue repair but also pathological fibrosis when chronically engaged.
Mitochondria, often dubbed the cell’s powerhouses, constantly face oxidative stress and potential damage, necessitating stringent quality control mechanisms such as mitophagy to maintain cellular homeostasis and function. Parkin-mediated mitophagy removes defective mitochondria, preventing the release of pro-inflammatory signals and limiting fibrogenic cascades. The novel findings illuminate how Hdac11-mediated repression of Parkin stymies this essential cleanup operation, precipitating an accumulation of dysfunctional mitochondria that skew macrophages toward an M2-type immune phenotype. This macrophage polarization fosters secretion of fibrogenic cytokines and growth factors, which in turn activate resident fibroblasts.
Notably, the study integrates in vitro cellular models with in vivo murine fibrosis models, allowing for a multi-layered understanding of Hdac11’s role in disease pathogenesis. Silencing Hdac11 expression revived Parkin-dependent mitophagy and reversed the fibrotic phenotype, substantially reducing myofibroblast accumulation and pulmonary fibrosis severity. Conversely, Hdac11 overexpression exacerbated lung fibrosis, underscoring its critical involvement. These functional validations highlight Hdac11 as a promising therapeutic target capable of interrupting the vicious cycle of macrophage-driven fibrogenesis.
The implications of this research extend beyond idiopathic pulmonary fibrosis, potentially influencing our understanding of fibrotic disorders across multiple organ systems where macrophage plasticity and mitochondrial dysfunction are similarly implicated. Epigenetic regulators like Hdac11 may represent a universal nodal point integrating environmental cues, immune cell state, and mitochondrial quality to dictate tissue remodeling outcomes. Targeting Hdac11 could thus offer a strategy not only for pulmonary fibrosis but also for cardiac, hepatic, and renal fibrosis—all conditions where myofibroblast-mediated scarring compromises organ function.
The authors also delve into mechanistic details elucidating how Hdac11 interacts with the Parkin promoter, impacting its transcriptional activation. Chromatin immunoprecipitation assays revealed Hdac11’s recruitment to specific regulatory regions of the Parkin gene, suppressing its expression via histone deacetylation. This epigenetic repression translates into diminished Parkin protein abundance, impeding mitophagic flux. Such mechanistic insights offer opportunities for designing small molecules or genetic tools capable of disentangling this interaction to restore mitochondrial quality control in fibrotic macrophages.
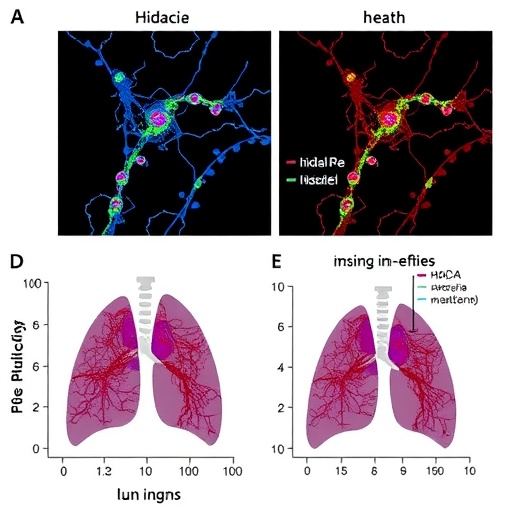
Furthermore, the research team evaluated the tissue localization and expression patterns of Hdac11 and Parkin in lung biopsies from IPF patients, confirming their inverse correlation and supporting translational relevance. Immunohistochemical analysis showed prominent Hdac11 expression within macrophage populations in fibrotic regions coinciding with suppressed Parkin levels, correlating with increased myofibroblast markers. These human data validate findings obtained from animal models and underscore the clinical potential of modulating the Hdac11-Parkin axis.
Mitophagy impairment has long been recognized as a contributor to chronic lung diseases, yet the regulatory nodes controlling this process have remained elusive. By identifying Hdac11 as a novel epigenetic brake on mitophagy, this study generates a paradigm shift in mitophagy-related fibrosis research. It prompts re-examination of how metabolic stress, epigenetics, and immune plasticity intersect to propagate fibrogenesis. Moreover, this refinement of fibrotic pathophysiology can guide precision medicine approaches that tailor treatment based on molecular subtypes of macrophage dysregulation.
Importantly, the study clarifies the dual role of macrophages in lung homeostasis and disease. While M1 macrophages classically promote inflammation and pathogen clearance, M2 macrophages are associated with immunosuppression and tissue repair. Dysfunctionally sustained M2 polarization, as orchestrated by Hdac11 via mitophagy inhibition, underlies maladaptive fibrotic remodeling rather than healing. This distinction is critical for therapeutic development since broadly suppressing macrophages risks impairing host defense, whereas modulating epigenetic and mitochondrial pathways offers specificity and reduces collateral damage.
The clinical portfolio for pulmonary fibrosis has expanded modestly in recent years with antifibrotic drugs such as pirfenidone and nintedanib, yet these agents slow but do not halt disease progression and come with substantial side effects. The elucidation of Hdac11’s role provides fresh opportunities for developing next-generation therapies that target the cellular and molecular origins of fibrosis. Epigenetic inhibitors or mitophagy enhancers could restore macrophage function and dampen fibrotic signaling with potentially improved efficacy and tolerability.
As the global burden of fibrotic lung diseases rises and survival remains dismal, this research injects renewed optimism. The integration of mitochondrial biology, epigenetics, and immunology as demonstrated here will likely inform future clinical trials and biomarker discovery efforts. Therapeutic manipulation of Hdac11 will demand rigorous preclinical testing to evaluate off-target effects given its nuclear functions, but the potential rewards are monumental—transforming the grim prognosis of IPF into manageable or even reversible fibrosis.
In conclusion, the seminal work led by Nie, Xu, and Liu marks a turning point in pulmonary fibrosis research by linking Hdac11-mediated suppression of Parkin-dependent mitophagy to macrophage M2-type polarization and myofibroblast proliferation. Their insights deepen our molecular understanding of fibrosis and open new therapeutic frontiers centered on epigenetic modulation and mitochondrial quality control. The scientific and medical communities eagerly await the translation of these findings into clinical strategies that can ultimately alleviate the suffering of individuals afflicted by this relentless lung disease.
Subject of Research: The role of Hdac11 in promoting idiopathic pulmonary fibrosis via macrophage M2-type polarization and myofibroblast accumulation through inhibition of Parkin-dependent mitophagy.
Article Title: Hdac11 promotes idiopathic pulmonary fibrosis through macrophage M2-type polarization and myofibroblast accumulation by inhibiting Parkin-dependent mitophagy.
Article References: Nie, Y., Xu, L., Liu, Y. et al. Hdac11 promotes idiopathic pulmonary fibrosis through macrophage M2-type polarization and myofibroblast accumulation by inhibiting Parkin-dependent mitophagy. Nat Commun (2026). https://doi.org/10.1038/s41467-026-71639-x
Image Credits: AI Generated

{kind=link}