A groundbreaking study recently published in Genes & Immunity uncovers a pivotal molecular mechanism behind the progression of pulmonary fibrosis, a debilitating lung disease with limited therapeutic options. Researchers led by Lv, Chen, and Zhou have identified that deficiency in alpha-calcitonin gene-related peptide (αCGRP) significantly exacerbates pulmonary fibrosis by promoting cellular senescence in alveolar type 2 (AT2) cells, the essential progenitor cells responsible for regenerating the lung epithelium. This discovery not only opens new avenues for deciphering the intricate pathogenesis of lung fibrosis but also introduces αCGRP as a potential therapeutic target for mitigating this relentless disease.
Pulmonary fibrosis is characterized by excessive scarring of lung tissue, leading to progressive respiratory failure. Despite advances in understanding the disease’s fibrotic cascades, effective clinical interventions remain elusive. The lungs’ alveolar epithelium, primarily composed of AT2 cells capable of both self-renewal and differentiation into alveolar type 1 cells, is crucial for maintaining lung integrity and function. The study underscores how senescence, a permanent state of cell cycle arrest commonly associated with aging and cellular stress, contributes to alveolar dysfunction and fibrotic progression.
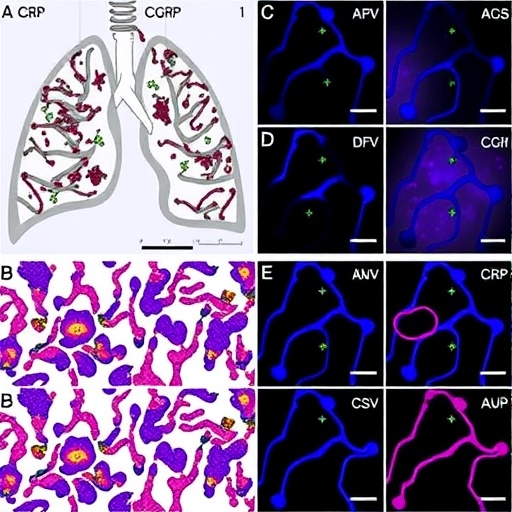
The research team employed a series of elegant in vivo and in vitro models to tease apart the role of αCGRP in modulating AT2 cell biology and fibrotic responses. αCGRP, a neuropeptide known for its vasodilatory and immunomodulatory functions, was found to exert a protective effect against lung fibrosis by restraining AT2 cell senescence. Mice deficient in αCGRP displayed enhanced fibrotic lesions after lung injury, accompanied by a marked increase in senescent AT2 cells, as evidenced by elevated expression of hallmark markers such as p16^INK4a and senescence-associated β-galactosidase.
Mechanistically, the study delineated how αCGRP orchestrates its anti-senescent effects by modulating intracellular signaling pathways pivotal to cell survival and proliferation. Loss of αCGRP disrupted these cascades, tipping the cellular balance towards premature senescence and apoptosis, which in turn impaired alveolar repair and fostered a pro-fibrotic microenvironment. This intricate crosstalk highlights the neuroimmune interface’s underappreciated role in lung pathology and raises provocative questions about systemic influences on local tissue remodeling.
A particularly striking aspect of this research is the link established between αCGRP deficiency and the senescence-associated secretory phenotype (SASP) in AT2 cells. The SASP, characterized by the release of pro-inflammatory cytokines, chemokines, and matrix remodeling enzymes, further amplifies tissue inflammation and fibroblast activation. Consequently, αCGRP-deficient mice showed elevated SASP factors, suggesting that αCGRP not only protects alveolar cells intrinsically but also tempers harmful paracrine signaling that accelerates fibrosis.
The implications of these findings are profound, as they suggest new molecular targets for intervention. Current antifibrotic drugs primarily aim to slow disease progression but do not address the underlying cellular senescence that drives tissue deterioration. By illuminating the neuropeptide’s critical regulatory role, this work advocates for therapeutic strategies that restore or mimic αCGRP signaling, potentially rejuvenating alveolar progenitors and halting fibrotic escalation.
Moreover, the study’s data hint at novel biomarker applications. Measuring αCGRP levels or detecting senescence markers in patient-derived AT2 cells might aid in early diagnosis or prognostic assessment of pulmonary fibrosis. Such biomarkers could personalize treatment approaches and monitor responses to emerging therapies targeting cell senescence pathways and neuroimmune modulation.
This study also invites exploration into how systemic factors such as neural signaling, inflammation, and aging intersect to influence lung disease susceptibility and progression. αCGRP’s role as a neuropeptide implicates the nervous system as a key player in maintaining pulmonary homeostasis, offering a fresh paradigm that transcends traditional inflammatory or fibrotic paradigms.
Future research inspired by these findings may investigate how manipulating αCGRP pathways affects other fibrotic conditions beyond the lungs. For example, liver or kidney fibrosis shares common molecular threads involving cellular senescence and chronic inflammation, raising the tantalizing possibility that αCGRP or related peptides could serve as broad-spectrum antifibrotic agents.
The translational potential of this discovery is underpinned by the established pharmacological profile of CGRP-related molecules. Already targeted in clinical settings for migraine treatment, these molecules could be repurposed or chemically optimized to treat pulmonary fibrosis—accelerating bench-to-bedside development.
In conclusion, the elucidation of αCGRP deficiency’s role in aggravating pulmonary fibrosis by promoting AT2 cell senescence represents a milestone in respiratory medicine. It enriches our understanding of the cellular and molecular dysfunctions driving lung scarring, offering hope for innovative treatments that restore lung regeneration capacity. As research advances, harnessing neuropeptide biology might transform the bleak outlook for pulmonary fibrosis patients, establishing new standards for diagnosis, prognosis, and therapy.
Subject of Research:
The role of αCGRP deficiency in promoting cellular senescence in alveolar type 2 cells and its impact on the progression of pulmonary fibrosis.
Article Title:
αCGRP deficiency aggravates pulmonary fibrosis by promoting senescence in alveolar type 2 cells.
Article References:
Lv, X., Chen, Q., Zhou, Z. et al. αCGRP deficiency aggravates pulmonary fibrosis by promoting senescence in alveolar type 2 cells. Genes Immun (2025). https://doi.org/10.1038/s41435-025-00361-3
Image Credits: AI Generated

{kind=link}